Freshly brewed drugs and newly branded devices frequently become available to the public. But before a product makes its way into a doctor's office or a drugstore shelf, it must be approved by the Food and Drug Administration (FDA) through one of two processes: 510(k) or PMA.
The Premarket Approval (PMA) is the FDA's most stringent review process, used for class III medical devices considered to "support or sustain human life, are of substantial importance in preventing impairment of human health, or which present a potential, unreasonable risk of illness or injury." PMA processes require scientific data and an extensive regulatory review before a drug can hit the shelves.
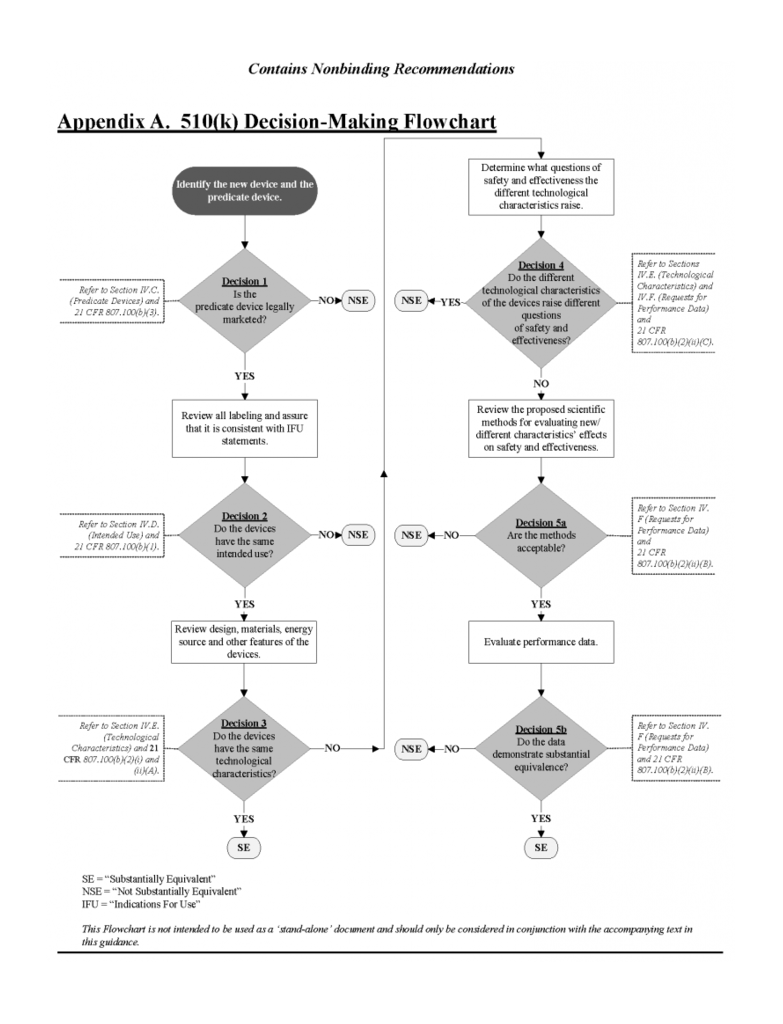
The 510(k) approval process, on the other hand, clears new drugs and devices for commercial distribution based on data from a comparable, or "substantially equivalent," product. The process "demonstrates that the device to be marketed is at least as safe and effective" as an existing class I, II or III device. Some items, like the majority of cosmetics, will be exempt from FDA review under the Federal Food, Drug, and Cosmetic Act.
The 510(k) approval process is a fast-track to the market. For manufacturers to gain approval through 510(k), there must be a "predicate" device approved after May 28, 1976 that illustrates substantial equivalence (SE). The FDA usually determines if the product has SE within 90 days based on the information submitted by the manufacturer.
According to the FDA, a device is substantially equivalent if, in comparison to a predicate it:
Though its been used to approve a large number of drugs and devices, the 510(k) program is highly controversial. The process expedites life-altering substances into consumer's homes without undergoing catered clinical trails. Some drugs come with serious side effects, and some devices inadvertently fail, leaving consumers to deal with disastrous side effects.
Consumer advocates believe that each drug and device approved for human use should undergo a strict, consistent, and comprehensive review. Manufacturers should be willing to drop chunks of change to research and develop the drugs they profit from.